





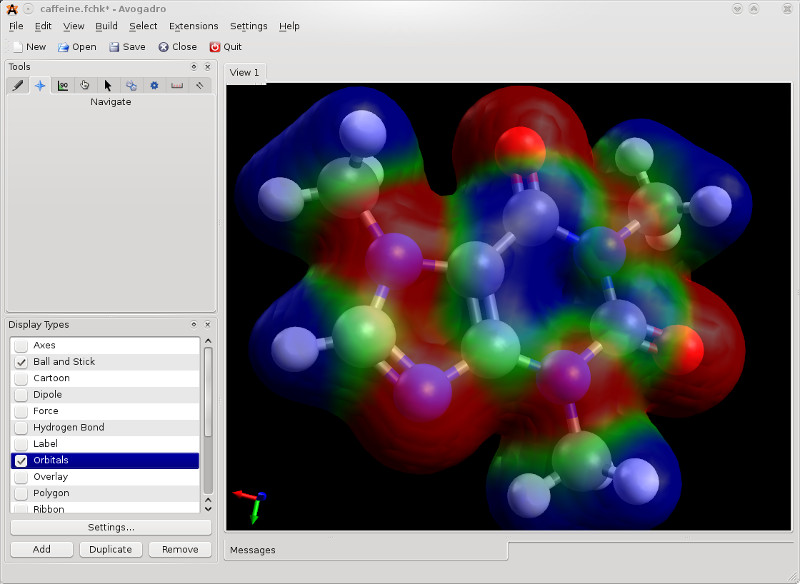
 молекулярне проектування і система моделювання спрямовані на молекули і біомолекули. Він може показати такі властивості як молекулярні орбіталі або електростатичні потенціали і можливості інтуїтивного молекулярного будівельника.
молекулярне проектування і система моделювання спрямовані на молекули і біомолекули. Він може показати такі властивості як молекулярні орбіталі або електростатичні потенціали і можливості інтуїтивного молекулярного будівельника.
Особливості:
✔Молекулярний модельєр з автоматичним силовим полем на основі оптимізації геометрії
✔Молекулярной механіки в тому числі обмеження і конформера пошуку
✔Візуалізація молекулярних орбіталей і загальних ізоповерхонь
✔Візуалізація коливань і побудова коливальних спектрах
✔Поддержка кристалографічних осередків
✔Ведіть покоління Gaussian, GAMESS і MOLPRO пакетів квантової хімії
✔Гнучка архітектура плагінів і сценаріїв Python
Avogadro підтримує формати файлів PDB, XYZ, CIF, Molden, а також Gaussian, GAMESS і створені MOLPRO.
Деякі з очікуваних можливостей:
Малювання (повузлове малювання; шаблони для поширених кілець; розширення загальних груп; малювання радикалів, зарядів, стрілок; підтримка кольору ...)
Редагування (необмежені можливості відміни і затримки; вирівнювання; масштабування; обертання (2D і 3D) ...)
експорт і імпорт (повністю підтримується експорт в формати SVG, OpenOffice.Org-Draw, EPS; базова підтримка імпорту та експорту CML1 і CML2).
Chemtool - 2D-редактор для малювання хімічних сполук під X11 (використовується GTK +). Він підтримує багато стилів з'єднання, більшу частину видів відображення тексту, що використовуються в хімії і сплайнові / дугові / криві стрілки.
Малюнки можна експортувати у формати MOL і PDB, для подальшого опису - в SVG або XFig, у вигляді малюнків PiCTeX, в растрові або Postscript файли (для деяких операцій використовується програма з XFig - transfig).
Також пакет містить допоміжну програму, cht, для обчислення формул і (точного) молекулярної ваги намальованого елемента файлу chemtool. Cht може викликатися прямо з Chemtool або з консолі.
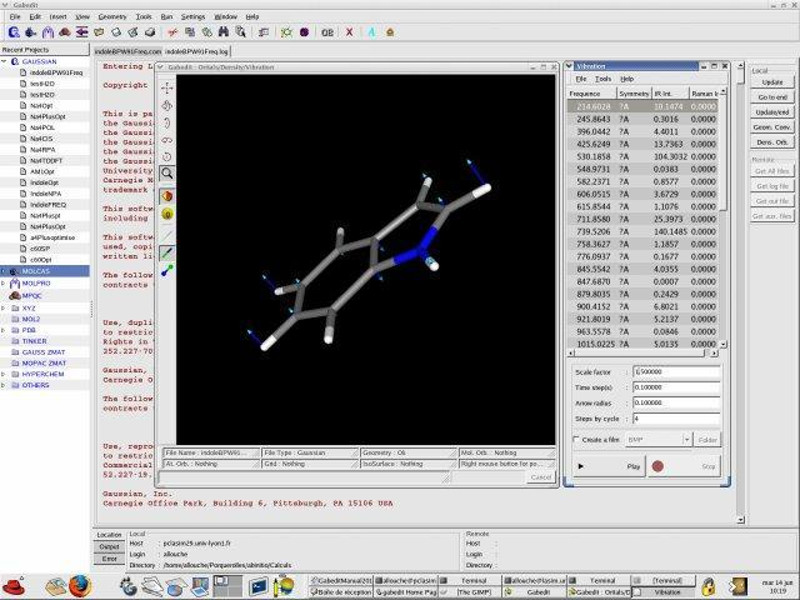 Графічний інтерфейс до обчислювальних хімічних пакетів Gamess-US, Gaussian, Molcas, Molpro, MPQC, OpenMopac, PCGamess і Q-Chem. Він включає тривимірний редактор молекул і вьювер
Графічний інтерфейс до обчислювальних хімічних пакетів Gamess-US, Gaussian, Molcas, Molpro, MPQC, OpenMopac, PCGamess і Q-Chem. Він включає тривимірний редактор молекул і вьювер
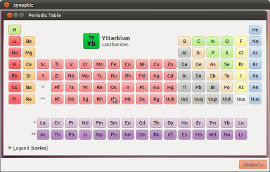
является периодическая таблица зрителя, который содержит подробную информацию о химических элементов.
Эта таблица показывает, которая позволяет элементами должен быть окрашен тематически несколькими свойствами, сортировки и просмотра списка элемента диалогового окна свойств, отображая различную информацию, в том числе исторические, термодинамические, электрохимических и кристаллографических свойств

Маленька програма, заснована на X / GTK +, дозволяє переглядати періодичну таблицю хімічних елементів і деяку, більш детальну, інформацію щодо кожного елемента. На даний момент доступно 118 елементів.
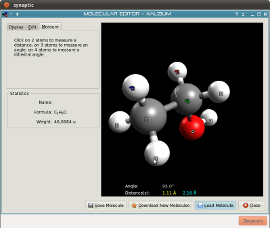
містить набагато більше інформації про періодичну систему Менделєєва, ніж будь-який студент університету коли-небудь хотів би дізнатися. Додаток також вирішує хімічні рівняння, показує зображення елементів і містить корисний словник термінів.
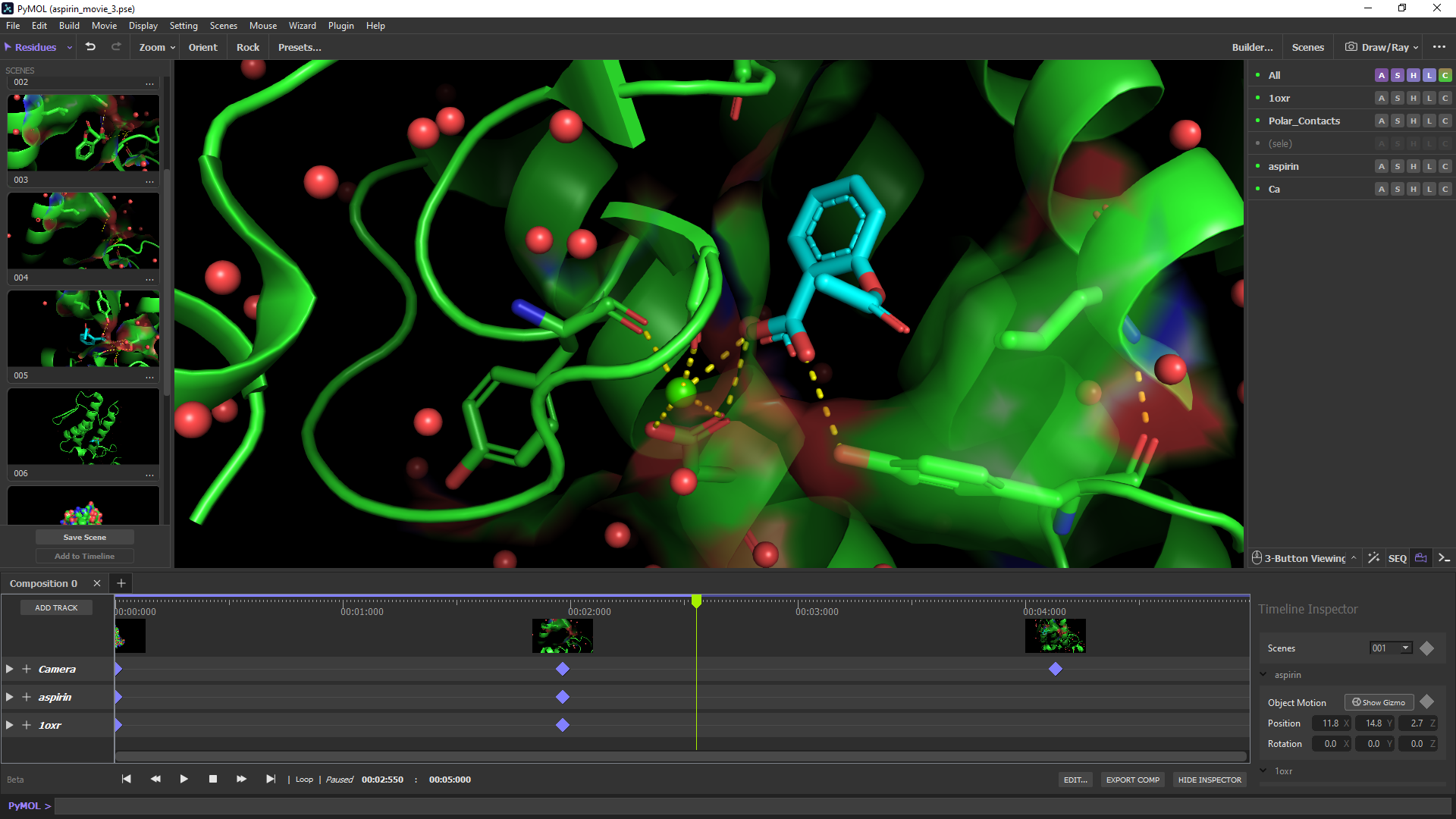 Це повністю готова система візуалізації молекул (молекулярної візуалізації) для використання в структурній біології.
Це повністю готова система візуалізації молекул (молекулярної візуалізації) для використання в структурній біології.
Дозволяє створювати високоякісні тривимірні зображення як малих молекул, так і біологічних макромолекул, насамперед білків.
Приблизно чверть усіх зображень структур білків, що публікуються в науковій літературі, зроблено за допомогою PyMOL.
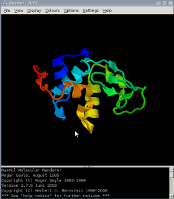 Комп'ютерна програма, призначена для візуалізації молекул і використовувана переважно для вивчення і отримання зображень просторових структур біологічних макромолекул, в першу чергу білків і нуклеїнових кислот.
Комп'ютерна програма, призначена для візуалізації молекул і використовувана переважно для вивчення і отримання зображень просторових структур біологічних макромолекул, в першу чергу білків і нуклеїнових кислот.
Починаючи з версій серії 2.7, RasMol поширюється по подвійний ліцензії (GPL або RASLIC). Тим самим, RasMol (поряд з Jmol і PyMOL) - одна з небагатьох програм візуалізації молекул з відкритим кодом.
RasMol залишається кращою з навчальних програм візуалізації молекул; крім того, не дивлячись на наявність більш складних аналогів, продовжує активно використовуватися молекулярними біологами та біоінформатики. Завдяки простій і логічній структурі призначеного для користувача інтерфейсу, програма проста для освоєння. Система команд ( "мова" RasMol'а) використовується в інших програмах, наприклад, в Jmol.
Вихідними даними для візуалізації служать координати атомів молекул (або комплексу молекул), що містяться в файлі формату Protein Data Bank (PDB). Файли з координатами атомів можуть бути скопійовані з одного із сайтів PDB.
Даний продукт багато в чому є аналогом (а подекуди і набагато більш функціональний) комерційного пакету Vector NTI, однак відрізняється кроссплатформенную, підтримкою можливостей сучасного заліза (многопоточность, CUDA) і можливістю самому писати плагіни або мишеклікабельно створювати обчислювальні ланцюжки з плагінів вже існуючих.
Основні можливості:
- Множина вирівнювання послідовностей на основі MUSCLE 3 і MUSCLE 4;
- Аналіз за допомогою прихованих марківських моделей, заснований на HMMER 2 і HMMER 3;
- Дизайн ПЛР-праймерів за допомогою Primer 3;
- Передбачення вторинної структури білків за допомогою GOR IV і PSIPRED;
- Філогенетичний аналіз за допомогою Phylip;
- Пошук сайтів рестрикції;
- Супершвидкий пошук простих і тандемних повторів;
- Аналіз сайтів зв'язування транскрипційних факторів на основі SITECON;
- Пошук відкритих рамок зчитування;
- Вирівнювання послідовностей за допомогою алгоритму з мита-Ватермана.
Програма для створення двовимірних малюнків молекул. Вона схожа за функціональністю на інші графічні програми, наприклад, ChemDraw.
Вона може читати і писати різні формати текстових і бінарних файлів, дає змогу обмінюватися інформацією між XDrawChem і іншими програмними додатками, а також може створювати зображення в популярних графічних форматах, таких як PNG і EPS.
ТОВ "УАЛІНУКС"
Написати: - у Viber - у WhatsApp - у Telegram
Зателефонувати: +380 (97) 33-55-1-88 (пн ... пт 10.00 - 17.00) (GMT+2)
E-Mail: [email protected]

![]()
![]()